UNDERSTANDING THE ROLE OF MDR FOR EU COMPLIANCE: A COMPREHENSIVE GUIDE TO MEDICAL DEVICE REGULATION
The European Union’s Medical Device Regulation (MDR 2017/745) represents the most significant overhaul of medical device legislation in decades. Since its full implementation in May 2021, the MDR has fundamentally transformed how medical devices are regulated, certified, and monitored throughout their lifecycle in the European market. Understanding the role of MDR in achieving EU compliance is crucial for manufacturers, distributors, and healthcare professionals navigating this complex regulatory landscape.
What is the EU Medical Device Regulation (MDR)?

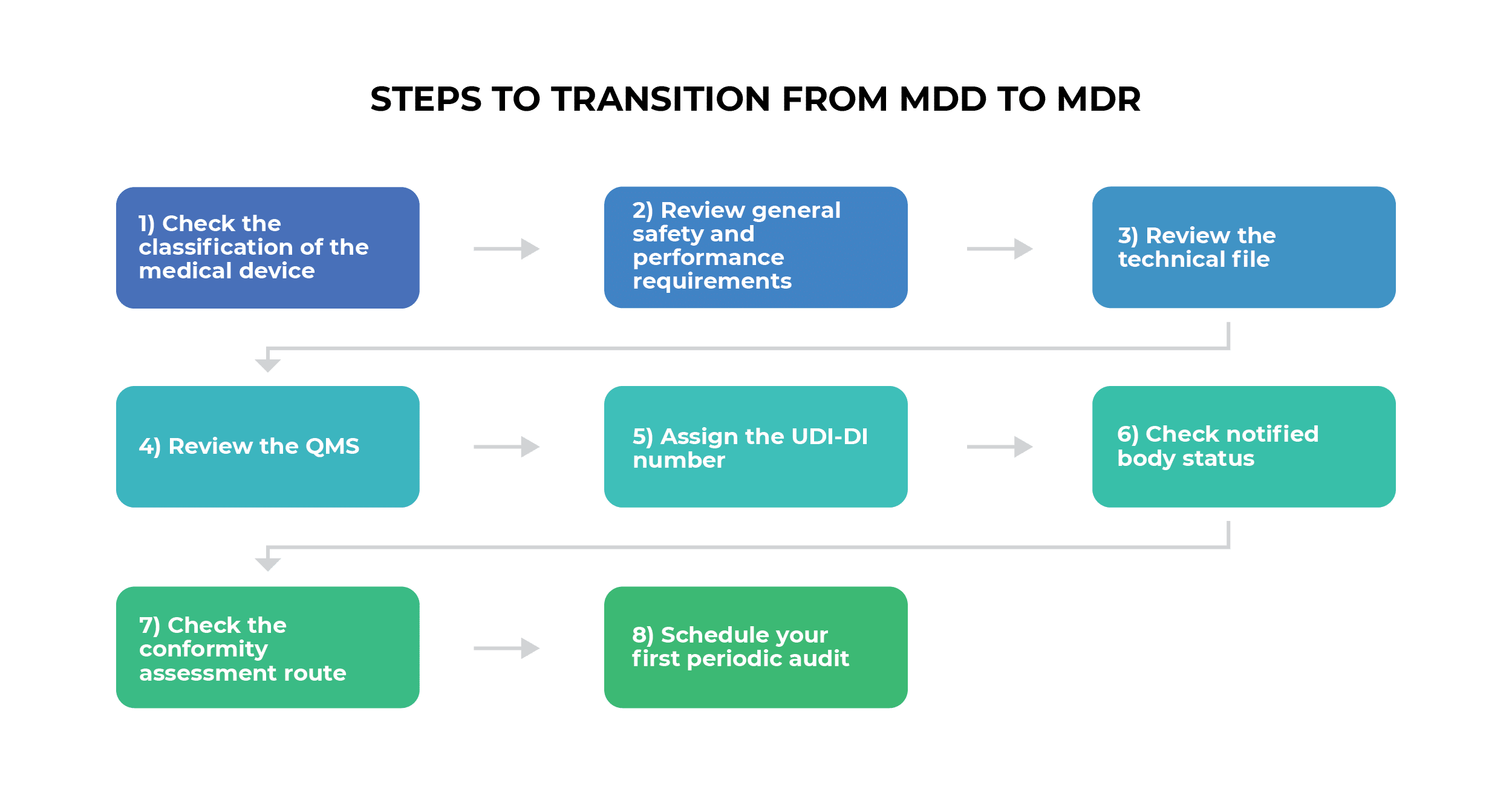
The Medical Device Regulation (EU) 2017/745, commonly known as the MDR, replaced the previous Medical Device Directive (MDD 93/42/EEC) and Active Implantable Medical Device Directive (AIMDD 90/385/EEC). This comprehensive regulatory framework was developed to address the rapid technological advancement in medical devices over the past two decades and to harmonize medical device regulation across all EU member states.
The MDR entered into force in May 2017 with a three-year transition period, becoming fully applicable on May 26, 2021. However, recent amendments have extended transition periods for certain devices to manage market disruption and ensure continuity of medical device supply. European Commission
Key Objectives of the MDR
The primary objectives of the MDR include:
- Ensuring patient safety and public health protection
- Supporting innovation while maintaining rigorous safety standards
- Creating a robust, transparent, and sustainable regulatory framework
- Enhancing post-market surveillance and vigilance systems
- Improving traceability through unique device identification (UDI)
Understanding MDR Compliance Requirements
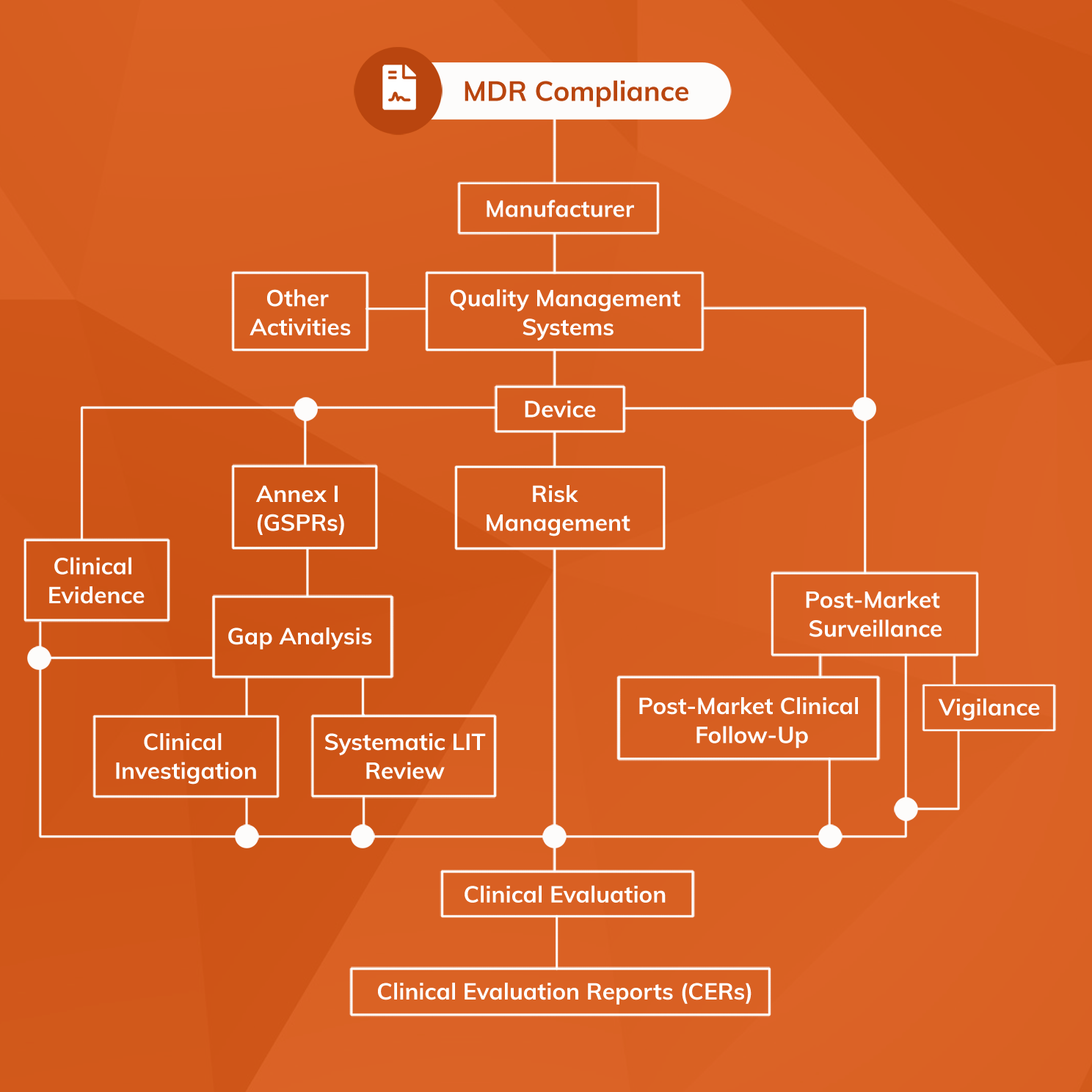
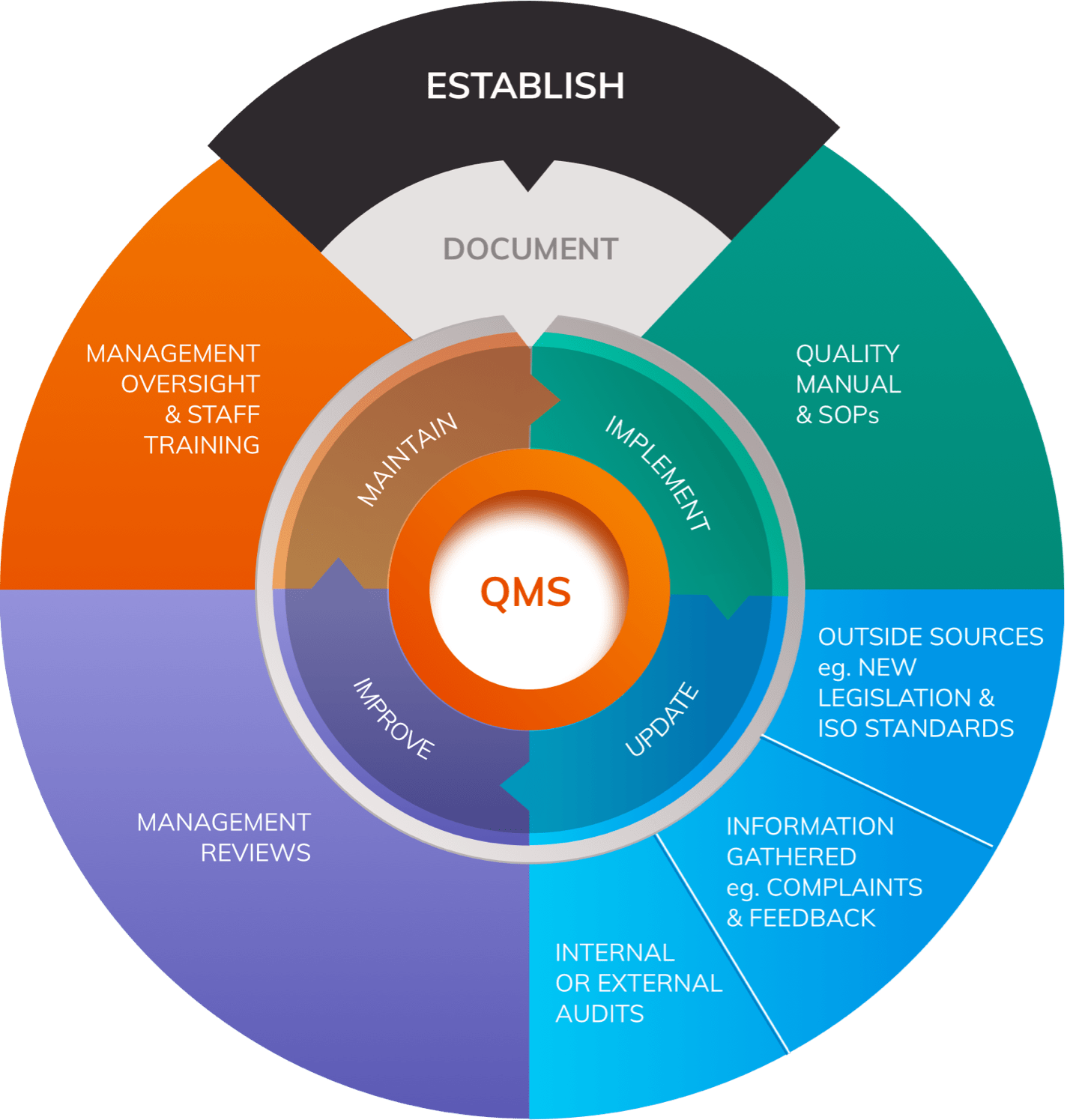
MDR compliance encompasses three fundamental domains that manufacturers must address comprehensively:
1. Medical Device Requirements
Medical devices must demonstrate:
- Suitability for their intended purpose
- Favorable benefit-risk profile
- Compliance with Annex I General Safety and Performance Requirements (GSPRs)
- Development within an appropriate Quality Management System (QMS)
2. Clinical Evidence Requirements
The MDR places unprecedented emphasis on clinical evidence, requiring:
- Comprehensive clinical evaluation for all device classes
- Robust post-market clinical follow-up (PMCF) systems
- Real-world evidence generation
- Continuous benefit-risk assessment
3. Regulatory Systems and Documentation
A comprehensive framework of systems and documents is mandatory, including:
- Quality Management System (QMS) per ISO 13485:2016
- Post-Market Surveillance (PMS) system
- Risk Management system per ISO 14971:2019
- Clinical Evaluation Reports (CER)
- Technical documentation per Annex II and III
- Person Responsible for Regulatory Compliance (PRRC)
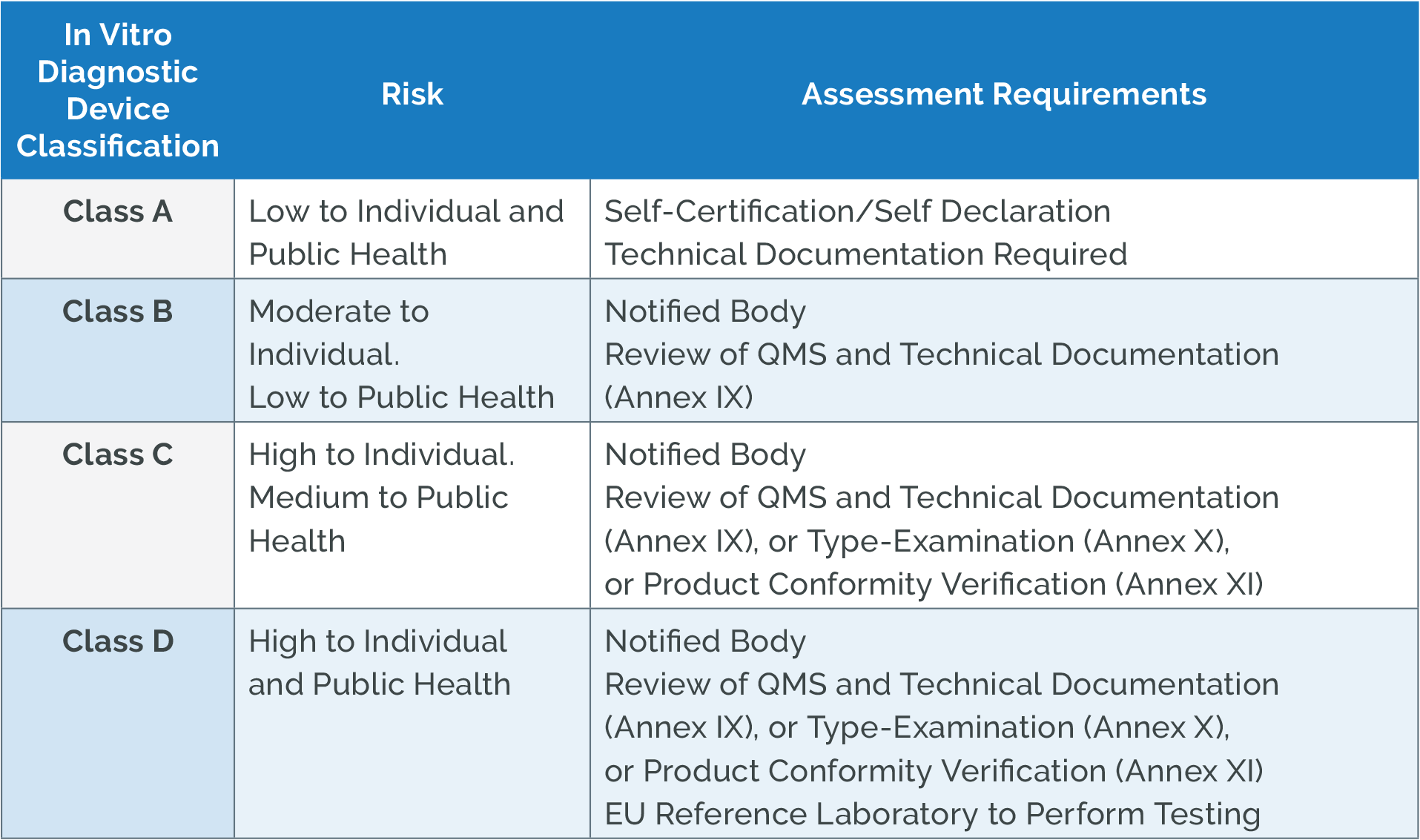
Medical Device Classification Under MDR

The MDR maintains a risk-based classification system with four classes: Class I, Class IIa, Class IIb, and Class III. Understanding this classification is crucial as it determines regulatory requirements and conformity assessment procedures.
Class I Devices (Low Risk)
- Non-invasive devices
- Generally self-certified by manufacturers
- Examples: bandages, examination gloves, wheelchairs
- Exceptions requiring Notified Body involvement:
- Sterile devices
- Devices with measuring function
- Reusable surgical instruments
Class IIa Devices (Low-Medium Risk)
- Short-term invasive devices
- Non-invasive devices contacting injured skin
- Examples: contact lenses, hearing aids, ultrasound equipment
- Require Notified Body assessment
Class IIb Devices (Medium-High Risk)
- Long-term invasive devices
- Active therapeutic devices
- Examples: ventilators, defibrillators, infusion pumps
- Require comprehensive Notified Body assessment
Class III Devices (High Risk)
- Life-sustaining or life-supporting devices
- Long-term implantable devices
- Examples: heart valves, pacemakers, artificial joints
- Most stringent requirements including Clinical Evaluation Consultation Procedure (CECP)
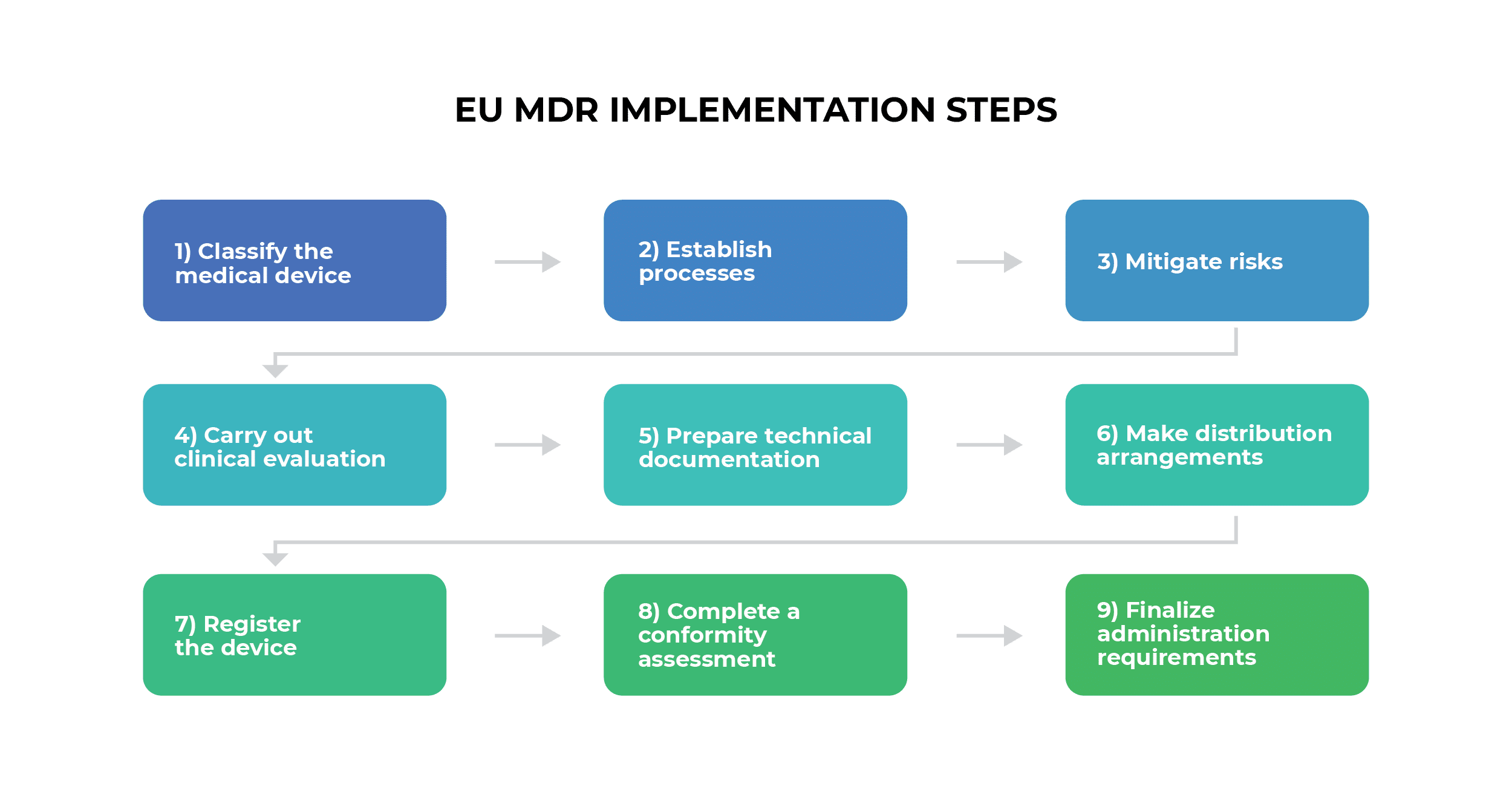
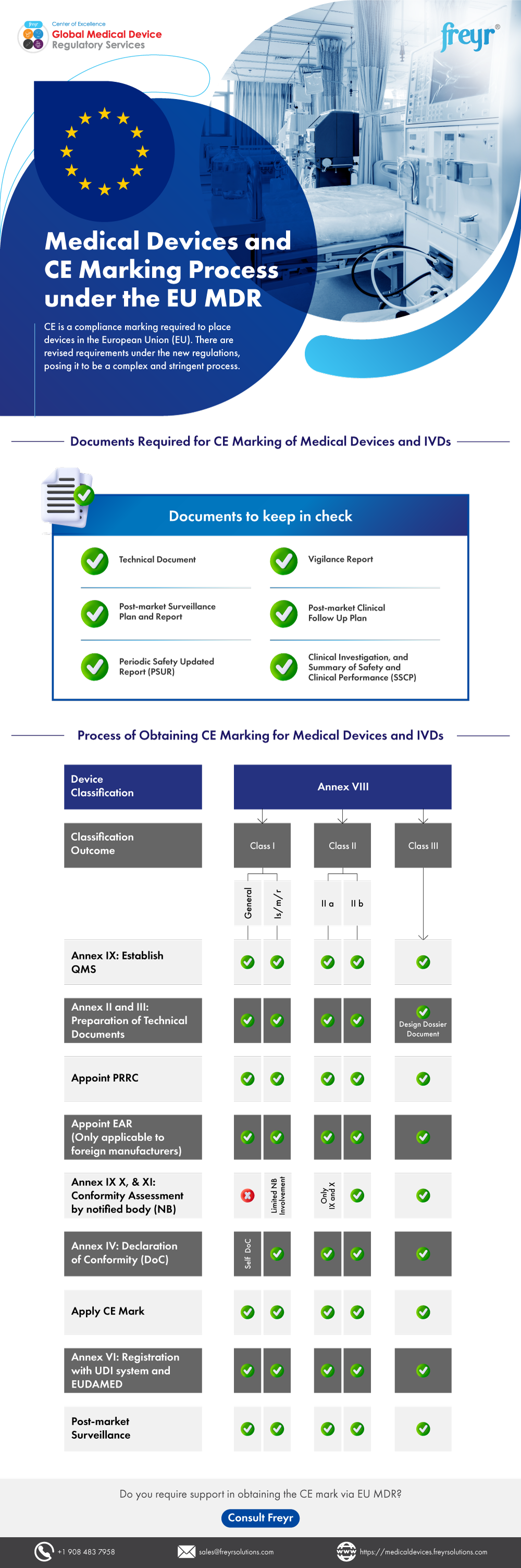
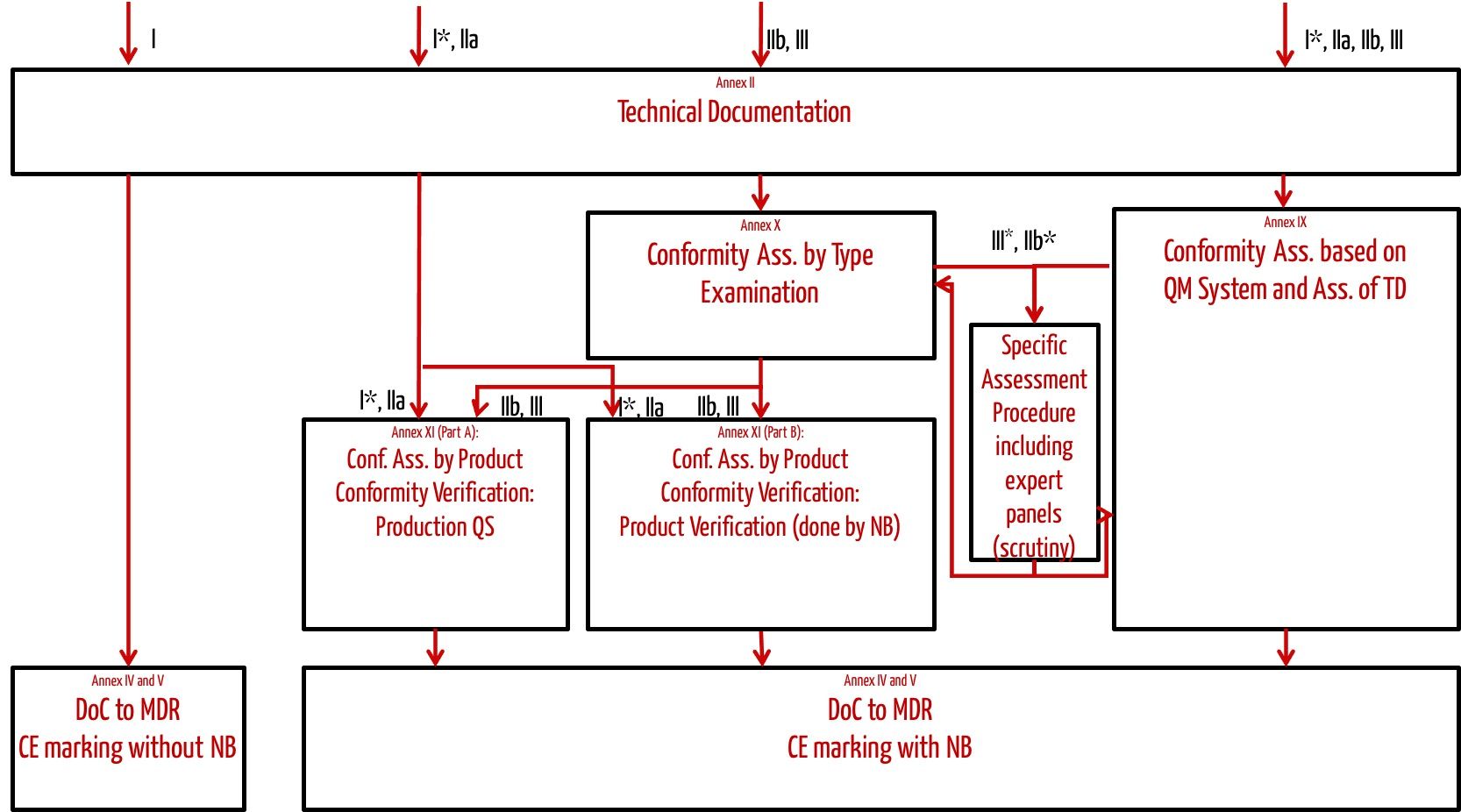
The CE Marking Process Under MDR

CE marking represents the manufacturer’s declaration that their medical device complies with all applicable MDR requirements. The process varies significantly based on device classification:
Class I Device CE Marking Process
- Device Classification: Determine appropriate classification using Annex VIII rules
- Technical Documentation: Prepare comprehensive technical file
- Declaration of Conformity: Self-declare compliance with MDR requirements
- CE Marking Application: Apply CE mark directly to device
- Registration: Register device and manufacturer in EUDAMED database
Higher-Class Device CE Marking Process

For Class IIa, IIb, and III devices:
- Pre-submission Preparation: Develop comprehensive technical documentation
- Notified Body Selection: Choose appropriate EU-designated Notified Body
- Conformity Assessment: Submit application for assessment procedure
- Technical Documentation Review: Notified Body evaluates all required documents
- QMS Assessment: Quality management system evaluation
- Certificate Issuance: Receive conformity certificate from Notified Body
- CE Marking: Apply CE mark with Notified Body identification number
- Market Placement: Device may be placed on EU market
Key Changes Introduced by MDR
The MDR introduced substantial changes that significantly impact compliance requirements:
1. Expanded Device Scope
The MDR’s broader definition now includes:
- Software as a Medical Device (SaMD)
- Devices without medical intended purpose (Annex XVI)
- Prediction and prognosis devices
- Nanomaterials and certain cosmetic products
2. Enhanced Clinical Evidence Requirements
- Mandatory clinical evaluation for all device classes
- Restricted equivalence claims, especially for Class III devices
- Comprehensive post-market clinical follow-up
- Summary of Safety and Clinical Performance (SSCP) for higher-risk devices
3. Strengthened Post-Market Surveillance
- Systematic post-market surveillance plans
- Periodic Safety Update Reports (PSUR)
- Enhanced vigilance reporting requirements
- Real-world evidence collection obligations
4. Unique Device Identification (UDI)
- Mandatory UDI system implementation
- Improved traceability throughout device lifecycle
- Integration with EUDAMED database
- Supply chain transparency enhancement
5. Person Responsible for Regulatory Compliance (PRRC)
- Designated individual with regulatory compliance oversight
- Specific qualification requirements
- Ultimate responsibility for MDR compliance
- Enhanced manufacturer accountability
The Role of Notified Bodies in MDR Compliance
Notified Bodies play a crucial role in MDR compliance for most medical devices. These EU-designated organizations conduct conformity assessments and issue certificates enabling CE marking.
Notified Body Designation Process
- Application: Organization applies to national authority
- Assessment: Competent authority evaluates capabilities
- Designation: Successful applicants receive designation
- Notification: European Commission notifies all member states
- Operation: Notified Body begins conformity assessments
Current Notified Body Capacity
As of 2024, approximately 50 Notified Bodies have gained MDR designation, a significant increase from the initial shortage in 2021. However, capacity constraints remain a challenge, making early engagement crucial for manufacturers. TÜV SÜD
EUDAMED: The European Database on Medical Devices

EUDAMED serves as the central database system supporting MDR implementation. This comprehensive platform enables:
Key EUDAMED Functionalities
- Actor Registration: Manufacturers, authorized representatives, and importers
- Device Registration: All medical devices placed on EU market
- Certificate Management: Notified Body certificates and declarations
- Vigilance Reporting: Serious incidents and safety notifications
- Market Surveillance: Competent authority oversight activities
- Clinical Investigation: Clinical trial registration and management
EUDAMED Rollout Timeline
Recent amendments (Regulation EU 2024/1860) introduced a gradual rollout approach:
- Phase 1: Actor and device registration (operational)
- Phase 2: UDI database (operational)
- Phase 3: Vigilance and market surveillance (gradual implementation)
- Phase 4: Clinical investigation module (future implementation)
Clinical Evidence and Post-Market Requirements
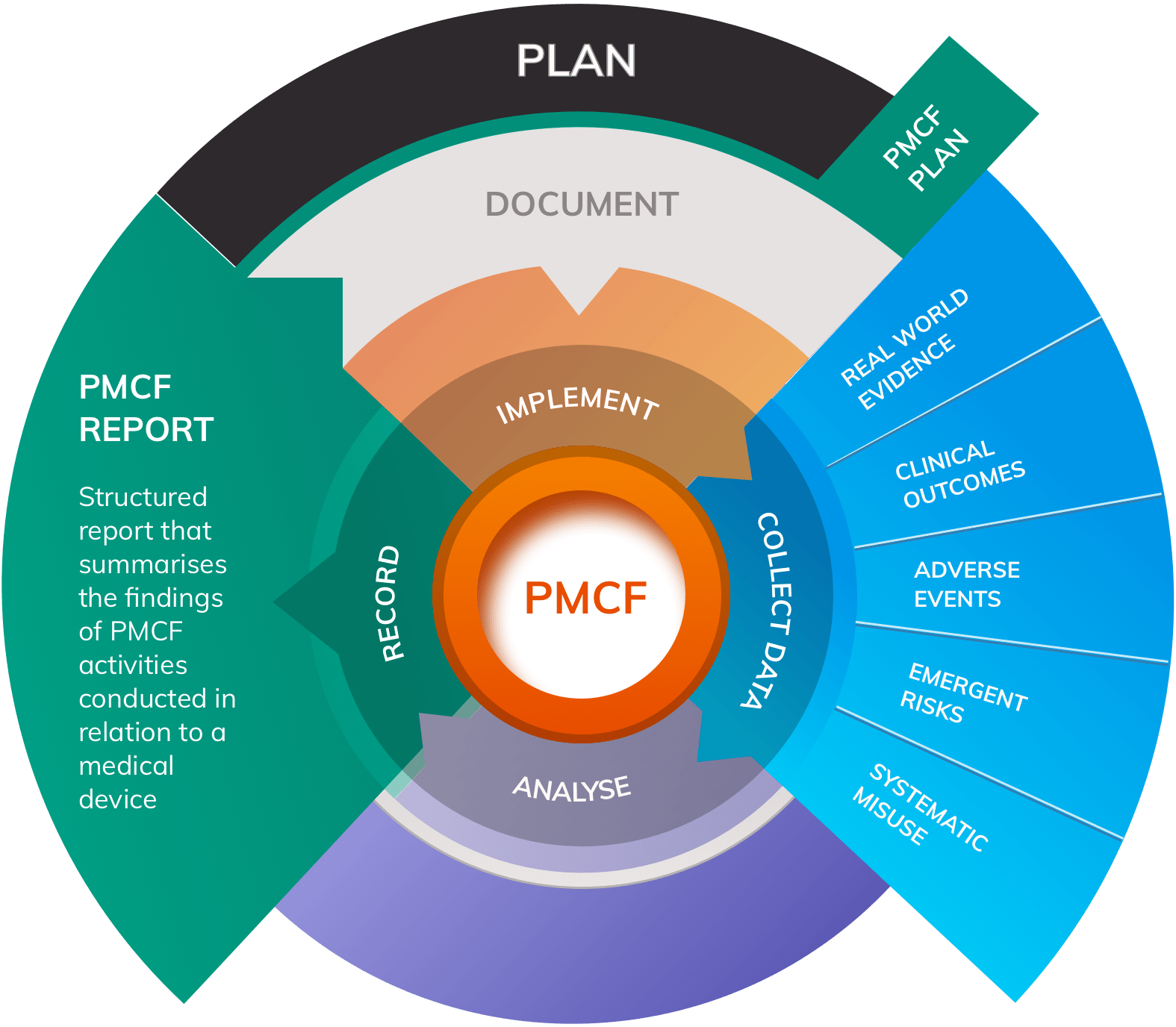
The MDR places unprecedented emphasis on clinical evidence throughout the device lifecycle:
Pre-Market Clinical Evidence
- Comprehensive literature reviews
- Clinical investigation data where required
- Equivalence demonstration (where applicable)
- Benefit-risk analysis
Post-Market Clinical Follow-up (PMCF)
- Systematic collection of clinical experience
- Real-world evidence generation
- Continuous benefit-risk assessment
- Safety signal detection
Clinical Evaluation Report (CER)
- Comprehensive analysis of all clinical data
- Regular updates based on new evidence
- Demonstration of continued compliance
- Support for device modifications
MDR Compliance Challenges and Solutions
Common Compliance Challenges
1. Resource Constraints
- Limited regulatory expertise
- Insufficient clinical evidence generation capabilities
- Budget constraints for compliance activities
2. Technical Documentation Complexity
- Extensive documentation requirements
- Integration of multiple regulatory systems
- Quality management system alignment
3. Clinical Evidence Generation
- Limited access to clinical investigation expertise
- Post-market surveillance system implementation
- Real-world evidence collection challenges
Strategic Solutions
1. Early Preparation
- Begin compliance activities well before deadlines
- Engage with Notified Bodies early in development
- Implement robust project management systems
2. Expert Consultation
- Partner with regulatory consultants
- Engage clinical evidence specialists
- Utilize Notified Body guidance services
3. Systematic Implementation
- Follow the establish-document-implement-update-improve-maintain cycle
- Integrate all regulatory systems effectively
- Ensure continuous monitoring and improvement
Recent MDR Amendments and Updates
Regulation (EU) 2024/1860
Recent amendments address:
- Extended transition periods for certain IVD devices
- Gradual EUDAMED rollout provisions
- Supply disruption notification requirements
- Streamlined implementation processes
Regulation (EU) 2023/607
Earlier amendments provided:
- Staggered MDR transition extensions
- Elimination of “sell-off” deadlines
- Enhanced flexibility for legacy devices
- Market continuity provisions
Future Outlook and Recommendations
Key Recommendations for Manufacturers
1. Proactive Compliance Approach
- Don’t wait for deadline pressure
- Begin compliance activities immediately
- Engage with regulatory experts early
2. Comprehensive System Integration
- Integrate all MDR requirements systematically
- Ensure quality management system alignment
- Implement robust documentation controls
3. Clinical Evidence Strategy
- Develop comprehensive clinical evaluation plans
- Implement effective PMCF systems
- Generate high-quality real-world evidence
4. Stakeholder Engagement
- Maintain regular Notified Body communication
- Participate in industry guidance development
- Stay updated on regulatory interpretations
Conclusion: Navigating MDR Compliance Successfully
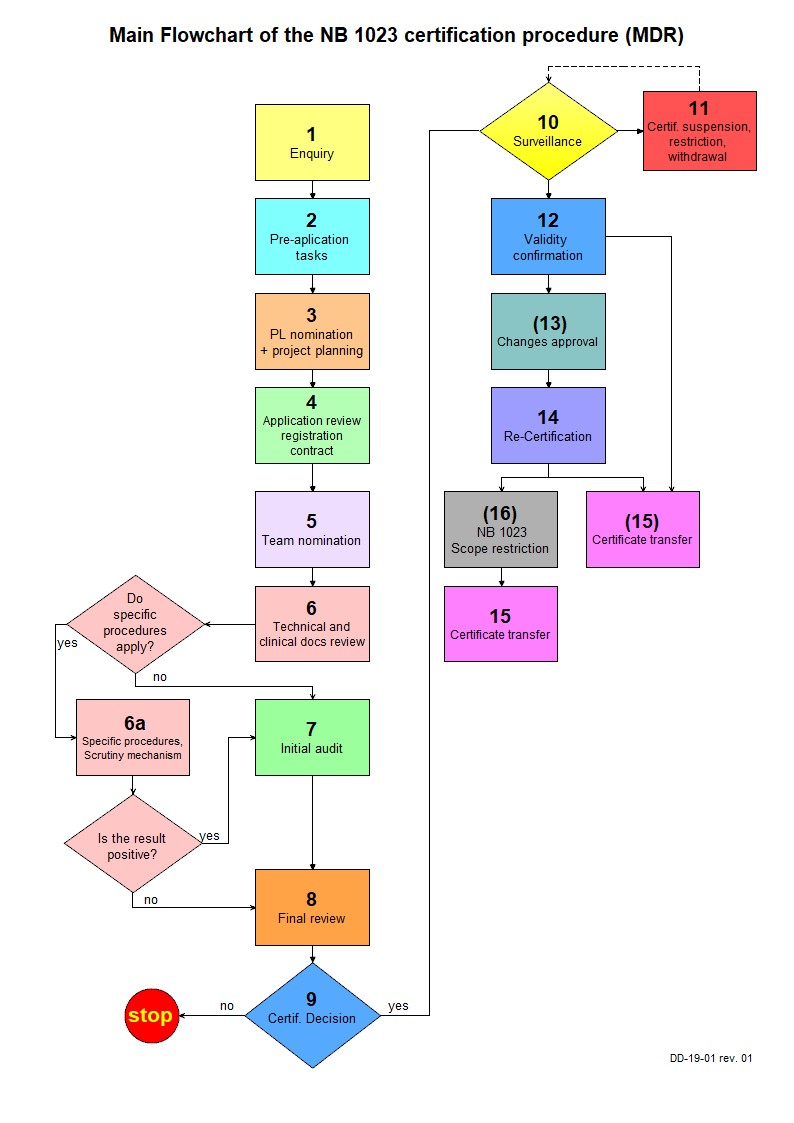
Understanding the role of MDR in EU compliance requires a comprehensive approach that addresses technical, clinical, and regulatory requirements systematically. The regulation’s emphasis on patient safety, clinical evidence, and post-market surveillance represents a fundamental shift toward more rigorous medical device oversight.
Success in MDR compliance depends on early preparation, systematic implementation, and continuous improvement. Manufacturers must invest in robust regulatory systems, clinical evidence generation capabilities, and ongoing surveillance activities to maintain compliance throughout the device lifecycle.
The MDR’s impact extends beyond regulatory compliance, driving innovation in clinical evidence generation, post-market surveillance, and device safety monitoring. Organizations that embrace these changes proactively will find themselves better positioned for long-term success in the European medical device market.
As the regulatory landscape continues to evolve with additional guidance documents, amendments, and implementation experience, staying informed and adaptable remains crucial. The MDR represents not just a regulatory requirement but an opportunity to enhance device safety, clinical effectiveness, and patient outcomes across the European healthcare system.
The journey to MDR compliance is complex but achievable with proper planning, adequate resources, and strategic implementation. By understanding the regulation’s comprehensive requirements and implementing systematic compliance strategies, manufacturers can successfully navigate this regulatory framework while continuing to bring innovative medical technologies to patients throughout Europe.